
Genomic-scale exchange of mRNA between a parasitic plant and its hosts.
科学家最近发现了一种潜在的新的植物沟通形式——寄生性植物菟丝子会与宿主植物交换大量的mRNA。
科学家最近发现了一种潜在的新的植物沟通形式——寄生性植物菟丝子会与宿主植物交换大量的mRNA。

Subtelny等研究人员发表了题为“Poly(A)-tail profiling reveals an embryonic switch in translational control poly(A)”的文章,通过PAL-seq (poly(A)-tail profiling by sequencing) 技术研究了胚胎发育时期poly(A)尾巴长度与翻译效率的关系。相关成果公布在2014年1月29日Nature杂志上。

mRNA的3’ UTR区是转录后调控的主要区域,虽然线虫的基因组已被解析,同时有大量的转录组数据支持,但截至到2011年大部分线虫mRNA的准确的polyA位点尚无准确注释。
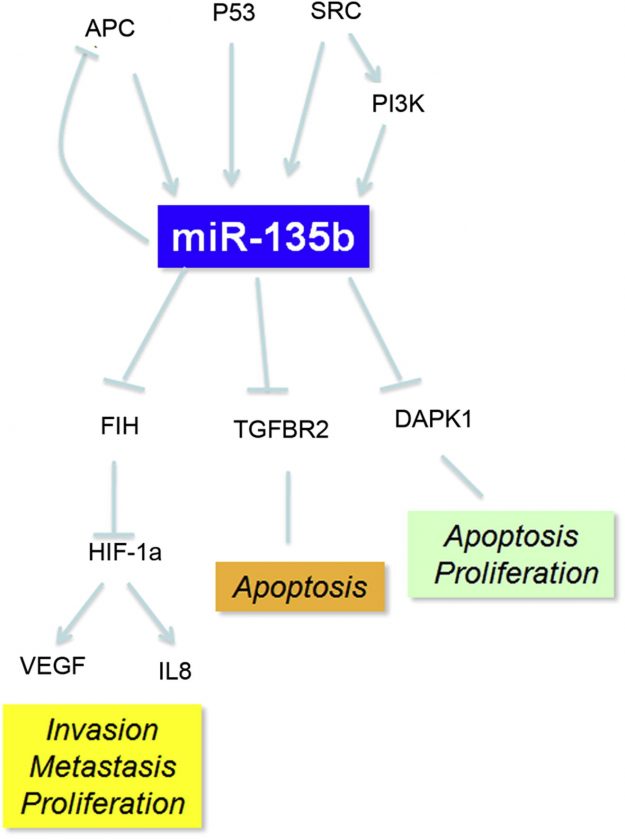
近期科学家在结肠癌研究邻域以“MicroRNA-135b Promotes Cancer Progression by Acting as a Downstream Effector of Oncogenic Pathways in Colon Cancer”为题发表了miRNA在癌症研究邻域的新进展。研究者首先采用microRNA-seq分别在两种结肠癌小鼠动物模型中检测了miRNA的表达差异,在两种动物模型中都检出miRNA-135b过量表达,最后的RT-PCR及原位杂交验证实验都证实了该结果。为了进一步验证动物实验的结果,随后研究者在62份临床样本中检测了miR-135b的表达量,另外还下载了392名CRC患者的miRNA-seq数据分析其中miR-135b的表达情况,在同一名患者的癌组织和癌旁组织中也能检测出miR-135b的显著差异。

最新关联宏基因组和宏转录组的一项研究 “Relating the metatranscriptome and metagenome of the human gut”,在线发表于2014年5月19日的《PNAS》杂志。这种技术是由美国Forsyth研究所、麻省综合医院和哈佛大学公共卫生学院等处的科学家开发的,该方法不需要专业人员和设施就可以收集用于基因组和转录组分析的唾液与粪便,同时还能保持样品的完整性;可减轻受试对象的一些负担,将使我们能够不受地理限制,开展纵向研究和大规模的收集研究这种方法也为消化道微生物组的基因组成和乳糜泻、口腔癌、牙周炎和肥胖症相关的细菌,提供了新的视角。

癌症的预测和早期诊断一直是医学界的难题,尽管已经有一些与癌症相关的基因异常得到鉴定,然而目前鉴定出来的与癌症相关的遗传性突变均定位于DNA的蛋白编码区,而且数目非常稀少。虽然全基因组范围内鉴定出了大量的非功能性突变,但是这些成果在医学诊断方面的用处并不大,其中一个可能的限制因素在于目前的研究主要局限于蛋白编码区的测序检测。

想要更加精确的对癌症患者进行诊断,就必须对肿瘤组织的基因组特征有更加清晰的了解,而该目的常常由于难以接近或获得肿瘤组织,或者能够得到的样品数过少使得研究者难以达到。收集循环系统中的肿瘤DNA或CTCs(circulating tumor cells)是一种比较有效的解决办法。但是使用CTCs进行测序分析的一个难点在于血液中存在的CTCs的数量非常稀少,因此构建文库的时候很容易出现过度扩增导致扩增偏向性和聚合酶错误。解决这个问题的一个思路是对同一个样本构建多个文库,然后对构建的文库进行统计学分析。研究人员运用该策略对前列腺癌的CTCs进行了测序分析。

部分前列腺癌对现有的抗激素治疗具有抗性,因此这部分前列腺癌成为了致命性疾病。现在,一项新的研究表明有一种方法或可用来治疗此类前列腺癌。